
FDA’s approach to evidence-based decision-making may not be addressed to the right people.
How can regulatory agencies encourage and coordinate high-quality data to improve regulatory decision-making?
The U.S. Food and Drug Administration (FDA) is grappling with that question in its real-world data and real-world evidence program. This program aims to use clinical data and evidence to inform regulatory decisions. FDA finalized these initiatives in 2018 under the 21st Century Cures Act of 2016, which sought to encourage innovation in developing medical products. With real-world data and evidence, FDA and the health care community can improve regulatory decision-making to design clinical trials, develop new treatment approaches, and analyze the risks and benefits of new medical technologies.
Last year, FDA updated its goals for use of real-world evidence to improve and expedite the drug approval process. More specifically, these goals include developing approaches for generating real-world evidence and using it to improve agency decision-making. The industry embraced this opportunity with the hopes it would be used correctly.
Although these goals may seem evenly split between industry and FDA, the responsibilities are not clean cut. When it comes to real-world data and evidence, FDA has issued guidance documents, which do not impose binding obligations.
This guidance-based approach works for FDA due to the nature of the cutting-edge technology. Guidance allows flexibility by shifting the responsibility for ensuring safety to researchers who can better assess their specific therapy. If researchers do not follow FDA guidance, they must prepare a validated procedure to explain why, and FDA still has the final say on whether that will be sufficient. Adequate validation requires documented evidence, at minimum, that the new alternative procedure is equivalent to or better than those in FDA guidance or, in rare cases, not applicable to the therapy at all.
Real-world data and evidence are slightly different. FDA’s framework and accompanying guidance on real-world data and real-world evidence appear to allow the use of data already provided to FDA. These submissions would allow data to be used in a different matter to allow FDA to make regulatory decisions, given that the researcher performed an analysis with the submission of applications for new drugs, investigational new drugs (IND), and biologics licenses.
FDA appeared to do this data review already in an unofficial capacity, but new guidance documents around real-world data and evidence would allow FDA to do so without waiting for researchers to publish their findings. FDA asked researchers to submit higher quality data to show that the researchers had conducted a more in-depth analysis with the understanding that FDA would use those submissions to guide regulatory decisions. There would presumably be little impact on both parties because, for the most part, the guidance was formalizing tasks that each was already doing.
These assumptions, however, were wrong. FDA can only move forward if it has consent to use data for a different purpose from the reason it was originally obtained. Although the 21st Century Cures Act waives the need for patients’ informed consent in the case of safety, intellectual property concerns remain. Without consent from researchers or a clear “go-ahead” from the U.S. Congress, FDA is back to the beginning with restricted use of data.
Instead, FDA guidance encourages researchers to submit real-world data to support safety changes, label changes, or marketing materials; or to submit a real-world evidence study design with the sources of data. With this interpretation, the guidance seems to require researchers to design a different study in addition to their ongoing drug studies to prove an update—which is already an FDA requirement.
This researcher-focused approach makes sense from an FDA standpoint.
According to the current process, researchers already conduct a validation to use as a preliminary study to start a real-world data and evidence investigation. The only additional steps are submitting a study design to use data from the validation and analyzing the resulting patient outcomes. In the eyes of FDA, data gathering is already part of the validation and researchers already observe patient outcomes, so the framework is expanding on the current role of the researcher.
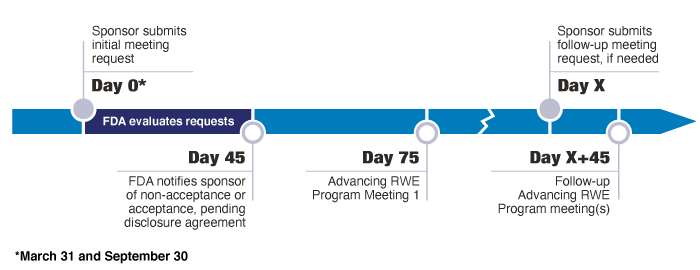
If FDA and researchers gather data at the time of setting up a pre-IND meeting, they can incorporate real-world data and evidence study into subsequent clinical trial design meetings. It would help support a researcher’s claim to use the validated method over FDA’s guidance. If researchers submit this information correctly, timely, and without any hiccups throughout the study, the use of real-world data and evidence can approve or update an existing application for new drugs and biologics licenses in a minimum of four months.
The problem lies mainly in the viewpoint of a researcher.
Currently, FDA offers no incentive to take additional steps when only a validation is required. Scheduling a meeting for a validation takes up to 30 days and approval may take another 30 days after submitting the data for approval. If researchers do not hear back from FDA within 30 days, they are free to implement the change. The researchers still need to conduct a thorough validation to show to FDA in the case of an audit, but there is a much more flexible and relaxed structure for a validation than for a complete study.
If researchers do not take additional steps for a complete study with real-world data and evidence, they can avoid the waste of resources. For those who have passed the pre-IND phase, these additional steps may also benefit their competitors more than themselves. If a study is successful, submitting the 12 line-item obligations and disclosure agreement of the extra study would enable competitors to start where the researcher’s real-world data and evidence investigation ends, instead of the researchers being the sole user of the validated method.
Even with FDA’s new goals for real-world data and evidence, this program is still an optional pathway with no convincing argument as to why a researcher would volunteer to conduct these studies. The real-world data and real-world evidence program is heading into its fifth year with only two approvals to show for it.
The only incentive to follow this new guidance is the potential loss of information. Researchers are aware and involved in the importance of real-world data and evidence on a grander scale. An issue, however, is that even if a researcher chooses to submit these documents for the greater good, it may not meet FDA’s criteria and would not move forward.
Because this real-world data and evidence program is voluntary, there is no motivation for the additional work.
If the new normal shifts, however, for researchers to submit high-quality, robust studies for every change to clinical trials and observational studies, then this change could promote greater efficiency. Researchers of a therapy will know their therapy best. If every researcher submits to that higher standard, FDA can compile and analyze the whole real-world data and evidence from every researcher, which is a clearer picture of safety than any single researcher could submit. FDA could then analyze that data to produce prompt, updated guidance and coordinate information.
The use of high-quality data and evidence to improve regulatory decision-making may evolve from the current program or from a different obligation, but it is necessary to achieve FDA’s goals most effectively.




{kind=link}