Responding to the coronavirus outbreak, a federal agency relaxes requirements on medical device manufacturers.
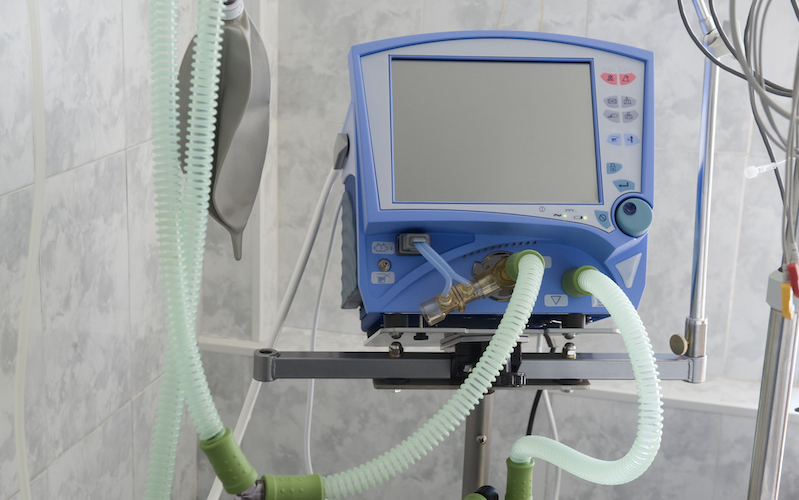
With intensive care units and emergency departments packed with COVID-19 patients and respiratory ventilators in short supply, the U.S. Food and Drug Administration (FDA) recently relaxed its regulatory enforcement efforts over the medical device industry.
Medical devices come in all shapes, sizes, and functions. Since medical devices treat, diagnose, or mitigate illnesses, many device manufacturers must submit an application to FDA called a Premarket Notification, or 510(k). FDA then reviews the 510(k) to ensure that the device is safe for use. Once deemed safe, the device can be marketed and sold.
But FDA review under 510(k) requires time. Considering the COVID-19 emergency, FDA issued guidance last week relaxing regulatory oversight in an effort to increase hastily the supply of respiratory ventilators.
All medical devices must be accompanied by a label based on data submitted in the 510(k) review process. Each label describes when, how, and under what circumstances for which FDA has approved the device. Any company found to be marketing or even overtly suggesting the use of a device beyond what the FDA-approved label denotes may have violated the law and could face legal action.
If a company changes the mechanics of a device, uses it to treat a different illness, or repurposes a device entirely, FDA normally requires additional data to be submitted that demonstrates the device is still safe and effective for use. With its recent guidance, FDA stated that “it does not intend to object to limited modifications to the indications, claims, functionality, or hardware, software, or materials of FDA-cleared devices…without prior submission of a premarket notification under section 510(k).”
FDA listed five device categories to which the guidance applies. Although these categories are not descriptive on their face, each medical device “cleared” for use by FDA is assigned a specific set of regulatory requirements by way of the regulatory category. For example, anesthesia machines designed and approved to deliver anesthetic gases are cleared for use under one regulatory category, while ventilators are cleared under another category.
In its guidance, FDA described four situations where modifications to the label of existing ventilator-like devices “would not cause undue risk” and may be made without submitting a new 510(k). Specifically, emergency ventilators and anesthesia machines can be repurposed to treat COVID-19 patients. Sleep apnea machines or ventilators approved for use by patients in their homes may also be modified and used for COVID-19 patients. Finally, devices which were originally approved to provide a concentrated flow of oxygen to patients are also within the scope of the new guidance.
In addition, FDA provided several examples of device modifications that it believes will not increase risks to COVID-19 patients using the modified devices. Such modifications included, among others, ventilator motor or power supply changes, the addition of extra filters, and changes to ventilator components that contact the patient during treatment.
For example, consider a European anesthesia device manufacturer. European electrical devices operate on a 240 volt power supply, but most U.S. outlets supply only 120 volts. Under FDA’s new guidance, if a European manufacturer wanted to modify its anesthesia machines to help meet the uptick in demand for standard ventilators, it would be permitted both to change the power supply to 120 volts as well as to repurpose the anesthesia machine into a standard ventilator without seeking prior FDA approval.
Another permitted change for anesthesia machines pertains to shelf life. Some device labels include a “duration of use” stipulation that can be thought of as the number of hours that any one machine can be used before requiring maintenance or replacement. If the modifications being made to anesthesia machines are within the scope of the guidance, FDA will not prevent those modified machines from being used longer than the time listed on the label.
FDA advises any manufacturer modifying a device to update the label to ensure that health care staff as well as patients are aware of the modifications. FDA suggests that manufacturers include a description of any modifications to the device, the intended use, and any potential risks associated with the modified device, as well as a clear distinction between what the device was originally approved for and how it is currently being used.
Although FDA’s guidance grants manufacturers significant leeway in making modifications, all changes should still be validated according to additional FDA guidance. Each manufacturer should test, record, and provide all validation data to the FDA upon request.
Furthermore, FDA encourages any manufacturer of a device in one of the categories listed in the guidance document that has not previously sold its device in the U.S. to submit an emergency use authorization (EUA) request to FDA. By offering EUAs to any manufacturer not previously marketing a device, FDA is providing ventilator device manufacturers a shortcut to get their products to those who need them most.
The authorizations outlined in this guidance were achieved through FDA’s careful evaluation and balancing of the risks that COVID-19 patients may face. If FDA did nothing to relax the regulatory hurdles that ventilator manufacturers typically face when modifying devices, an unknown number of COVID-19 patients would have died simply because ventilators were unavailable.
Death is always an unacceptable outcome in risk management and requires a reevaluation of the approach being taken.
FDA’s recent guidance document continues the preemptive approach that the agency has pursued in the face of the COVID-19 crisis. Historically, FDA has been mostly reactive: establishing or modifying policies or regulations only after significant harm has occurred. The approach taken in FDA’s recent guidance—although narrow in scope and only applicable during the COVID-19 crisis—reflects an agency taking unprecedented steps in an attempt to keep Americans safe in the most uncertain of times.


